BIO
I am a Research Software Engineer at the University of York, a position I’ve been in since March 2023. I am currently a part of the PlasmaFAIR project in the York Plasma Institute, where we perform health-checks on community code to improve the sustainability of plasma software. Additionally, as part of the Condensed Matter Dynamics group, I have extended community codes to better understand the material properties of solids.
I completed my DPhil at the University of Oxford, where I investigated the influence of quantum mechanical effects on solid-state nuclear magnetic resonance parameters. At the same institution I obtained my MSc in Theoretical and Computational Chemistry, and at the University of Warwick, I obtained my BSc in Chemistry.
I am particularly interested in the quantum mechanical nature of materials. I like to understand why electrons interact the way they do, and how we can use that understanding to better predict novel materials for practical applications. I like to place a strong emphasis on the sustainability of software, to ensure the community stays up-to-date and the research output is accessible to all and reproducible.
PROPOSAL
Value to the RTP and Upskilling Trajectory
This project focuses on the Explore stream, as the applicant already has a basic understanding of the mathematical and algorithmic principles of machine learning (ML) techniques. This knowledge has been applied through small, self-directed projects involving the implementation of neural networks primarily for learning purposes, rather than in a collaborative production environment. The project will aid in developing skills on the practical application of modern AI workflows for scientific research, including best practices for data handling, performance considerations and confidence tests to attain the validity of results. This upskilling will position the applicant to contribute effectively to real-world scientific challenges within the community, both during this project and beyond.
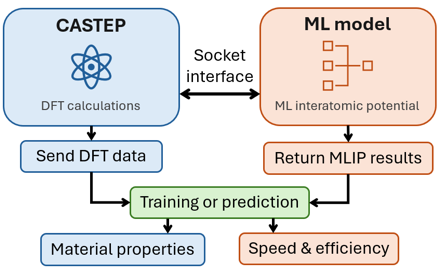
Scientific and Technical Workplan
The CASTEP electronic structure package is a leading electronic structure code that calculates the properties of materials from first principles methodologies. It implements density functional theory (DFT) to simulate a variety of atomic and material properties. To build on and apply skills gained through the training programmes and mentorship, a small prototype AI assistant for CASTEP will be created as an initial illustrative project. This will be able to create input files and scripts, as well as provide information to users on the programme’s functionality. An open source model, such as Llama or Gemma, will be used as a ML model foundation.
A key limitation in current materials modelling is the trade-off between the accuracy of ab initio methods and the computational cost required to compute physical properties at scale. One solution is the use of machine learned interatomic potentials (MLIPs). Using a socket interface within CASTEP, an MLIP can be trained that reads DFT computed electronic structure data to output material properties. These potentials can make use of the accuracy that DFT provides, but at a cheaper computational cost. See Phys. Rev. X 8, 041048. Currently, the spin property of the electronic density is not accounted for in the ML model, meaning the results lack applicability to magnetic materials. The proposed project will aim to extend the present scheme to make use of the electron spin, which will improve the ML model predictions.
Furthermore, the MLIPs are trained on the atomic forces. The proposed project will also consider the effect of training the ML model on the Hessian, that is the derivative of the forces. One advantage of this is the natural connection to vibrational properties, such as phonons, which are a function of the Hessian. Confidence tests can be made to calculate the zero-point energy, which would usually require expensive path integral molecular dynamics calculations to attain. Additionally, the geometry optimisation routine utilises the Hessian to calculate the next-step direction in the algorithm. Employing an MLIP trained on the Hessian may improve the accuracy and speed of these calculations, which are a vital step in almost all material studies.